-
Americas

-
Asia & Oceania

-
A-I
J-Z

EMEA Thought Leadership
Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more -
Middle East & Africa

EMEA Thought Leadership
Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more
Regions
-
Americas
-
Asia & Oceania
-
Europe
-
Middle East & Africa
-
Americas
-
Asia & Oceania
-

Europe
Europe
- Adriatic
- Belgium
- Bulgaria
- Czech Republic
- Deutschland
- España
- France
- Greece
- Hungary
- Ireland
- Israel
- Italia
EMEA Thought Leadership
Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more -
Middle East & Africa
EMEA Thought Leadership
Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more
Our Latest Whitepapers
Get the latest insights on our life sciences, healthcare, and medical technology solutions in Asia Pacific.
Learn MoreBlogs, Case Studies & Podcasts
Explore our library of insights, thought leadership, and the latest topics & trends in healthcare.
Discover Insights
IQVIA APAC Insight
"A curation of IQVIA's best thinking on topics and trends driving change, disruption, and progress in the Asia Pacific healthcare market.
Read The Latest Issue
Career Opportunities
Improving human health requires innovative thinkers who are willing to explore new ideas and build on successes. Unleash your potential with us.
Search Jobs
Blog
Overview of Companion Diagnostics and its Regulatory Trends in Asia Pacific
Aug 03, 2023
The advent of precision medicine has launched the trend towards individualised treatments, offering a glimpse into the future of healthcare1. These methods leverage on technology to tailor healthcare treatments according to individual patients’ genetic biomarkers (e.g., The presence of a mutation in a specific gene) to increase the benefit-to-risk ratio of treatment2. As such, Companion Diagnostics (CDxs) are a class of medical devices that play a key role in this transition from generic to individualised treatment.
Introducing CDx
Companion diagnostics are tests or assays that help identify patients who are most likely to benefit from a particular treatment or medication. These tests are often used in precision medicine to tailor treatments to individual patients based on their specific characteristics, such as genetic makeup, biomarker expression, or other diagnostic criteria.
Many regulatory agencies, such as the U.S. Food and Drug Administration (FDA), require companion diagnostic tests for the approval of certain targeted therapies. Knowing companion diagnostic values is essential for obtaining regulatory approval for these treatments.
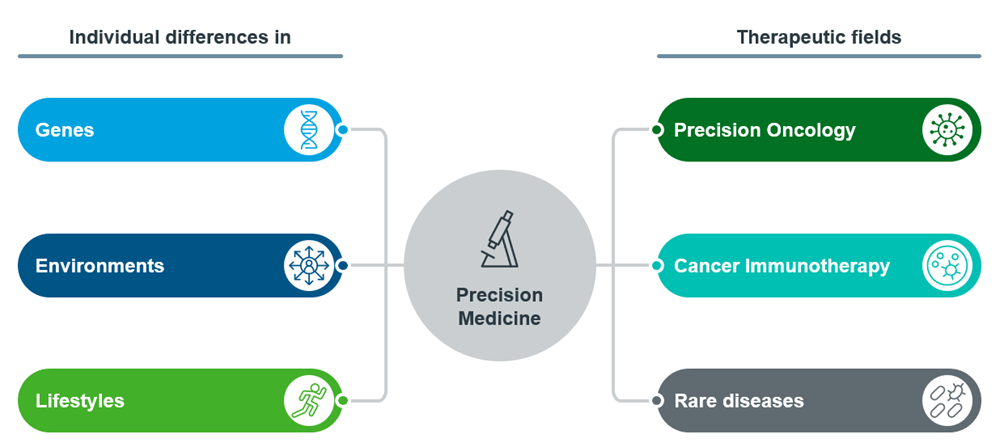
Figure 1: Overview of Precision Medicine
“Companion Diagnostic” is an aptronym for the class of devices it describes -- Companion, in that it is often developed alongside a specific therapeutic drug; Diagnostic, in that it is a biochemical assay that determines if the patient is likely to benefit from treatment.
CDxs can serve 3 main purposes3, to identify any of the following: Genetic subgroups that have a higher likelihood of benefitting from a specific therapeutic; patients at a higher risk of serious side effects from treatment with a specific therapeutic; and any adverse responses to modify treatment to increase its suitability and safety.
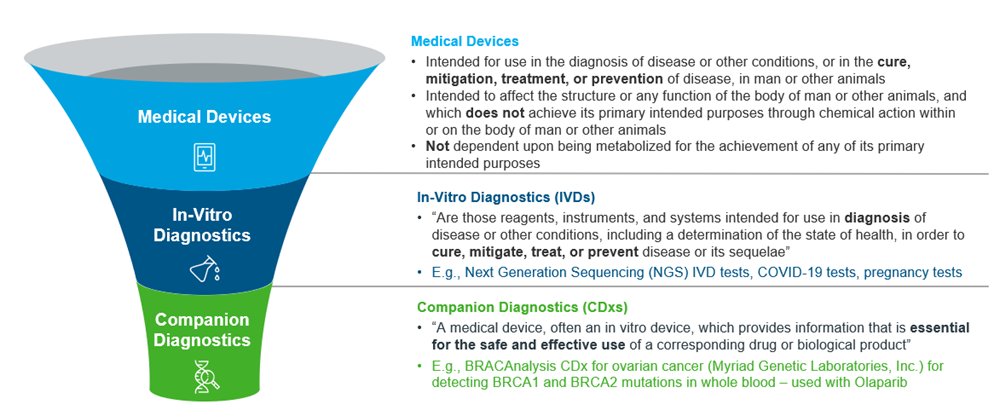
Figure 2: Overview of Medical Devices, IVDs and CDxs, adapted from US FDA
CDxs accompany and expedite the process of showcasing its corresponding drug’s effectiveness in the specific population most likely to benefit from it, in order to highlight the drug’s efficacy. Predictive biomarkers are key in the successful implementation of precision medicine4, and the number of US FDA approved CDx has steadily increased ever since the introduction of the drug-companion diagnostic co-development model in the late 1990s.
A brief history of CDx: Case study of Herceptin
The term “companion diagnostic” was first introduced in the late 1990s – with the pivotal point being the simultaneous approval5 of Herceptin (trastuzumab) and its corresponding CDx, HercepTest™. The parallel development of this monoclonal antibody targeting HER2 overexpression, along with its corresponding immunohistochemical assay (IHC) identifying its target as a biomarker6 has since welcomed a new era for cancer drug development. Due to the targeted mechanism of action of Herceptin by binding to the HER2 protein (whose expression levels vary across individuals), it was a critical challenge to identify the patient subpopulation who was most likely to benefit from its treatment.
Genentech (Herceptin’s developer) used a clinical trial assay (CTA) to select for HER2 positive patients during its clinical development, and HercepTest™ was an optimised IHC assay that was developed by Dako during Herceptin’s phase III trials. Since HercepTest™ showed concordance with this CTA, Herceptin and HercepTest™ were simultaneously approved by the FDA in September 1998.
HercepTest™ was vital to the market authorisation of Herceptin. Alternative sample size calculations published in Clinical Cancer Research based on the data from the phase III trials of Herceptin in metastatic breast cancer showed that had the 469 patients not been pre-screened for HER2 overexpression, the number of patients that would have been required to demonstrate the same statistical difference between treatment arms would have been over 80005.
Former ASCO president Gabriel Hortobagyi echoed the importance that CDxs played in the success of Herceptin by stating in Seminars in Oncology: “If an assay did not exist to identify the patient population likely to respond to therapy, trastuzumab might have been discarded during development because of insufficient activity in an unselected patient population.”5
Furthermore, the benefit that CDxs bring to the therapeutics they accompany even reaches beyond their main purpose of spearheading regulatory approval as CDxs contribute further to the actual uptake of the therapeutic they accompany. Herceptin was initially not recommended for general gastric cancer treatment2 in the UK, and only received a favourable recommendation when HercepTest was able to identify the subgroup of cancer patients that overexpressed the HER2 protein target of Herceptin.
CDx trends in oncology
Precision oncology has been a burgeoning field as scientists seek to better understand the specific pathophysiology of different types of cancer to reduce off-target side effects7. As such, it is important to identify the unique biomarkers of cancer patients that may predict their response to treatment. It is therefore of no surprise that almost all approved CDxs are indicated for use with cancer therapeutics, as opposed to other fields.
Traditionally, therapeutic products that accompany CDxs in oncology involve a single drug, although the US FDA has encouraged the use of CDx with a specific group of related drugs targeting the same mutations, to maximise the benefit that CDxs bring to patient treatment. The labelling of US FDA-approved CDx for a group of related drugs (i.e., for patients with the same mutation(s) instead of individual drugs) reduces the need for clinicians to order separate CDx that identify the same mutation (e.g., EGFR mutations of NSCLC). This minimises the turnover time for the patient to receive appropriate treatment. Although, the US FDA has established this guidance with the caveat that clinical evidence must be developed for at least one device, for the same specimen type, for each drug.
In fact, as much as customising treatments to patients has increased success in oncology treatments for drugs with a CDx, the pace at which such drugs develop at can also paradoxically present as a challenge, as corresponding CDxs may not be implemented fast enough8 to match the speed at which new therapeutics enter the market.
In 2020, a large-scale study of newly diagnosed advanced NSCLC patients in the US found that ~23% of patients did not receive genomic testing for any of four guideline-recommended therapeutic targets (ALK, BRAF, EGFR, and ROS1 alterations) before first line treatment8. This is problematic as NSCLC mutations are not homogenous across all patients, and most advanced NSCLC patients (~65%)9 have the potential to receive treatment that targets their specific genomic alterations. First-line treatment for patients with the mutations in the aforementioned targets should have been customised to their unique mutations.
Recent trends in CDx
As CDxs have gained traction, their regulations have also evolved over the years. For an overview of how this evolution has materialized, one can look to the evolution of regulations for CDxs in the U.S., where the first final guidance was issued in 2014. Even now, regulations are still evolving in the U.S. such as obtaining broader labelling. Additionally, the U.S. FDA is also piloting a program for oncology-specific IVDs.
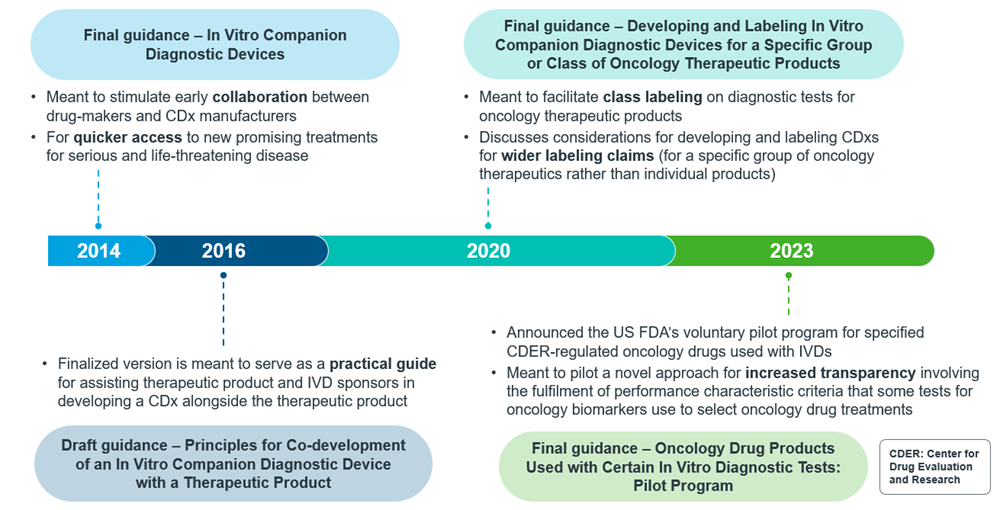
Figure 3: Timeline of CDx guidance publication from US FDA
In order to navigate the US FDA regulations around broader labelling for CDxs, such as use with a specific group of oncology therapeutics, manufacturers are required to justify this with valid scientific evidence under appropriate requirements for approval and clearance. This justification includes the demonstration of analytical and clinical validity10.
To achieve analytical validity, new CDxs need to demonstrate that the CDx can perform as intended in terms of sensitivity, specificity, accuracy, precision, and other relevant performance characteristics using a specified technical protocol, across the range of biomarkers that inform its indication, in addition to the specific biomarkers that the CDx itself is detecting.
It is also important to consider clinical validity. Differences in the diagnostic thresholds of different CDxs could present as one of the challenges in broadening the labelling of CDxs for oncology treatment. Since each CDx has a unique algorithm to qualify a sample for treatment, these differences make it difficult to ensure concordance between CDxs analyses and CTAs. Manufacturers should also refrain from modifying the thresholds of their CDxs just to fit the samples that qualify under their CTAs, as this counterintuitively reduces the chances of gaining market authorisation11.
The figures below show the general workflow for bringing a CDx to the market in the US (Figure 4) and the EU (Figure 5):
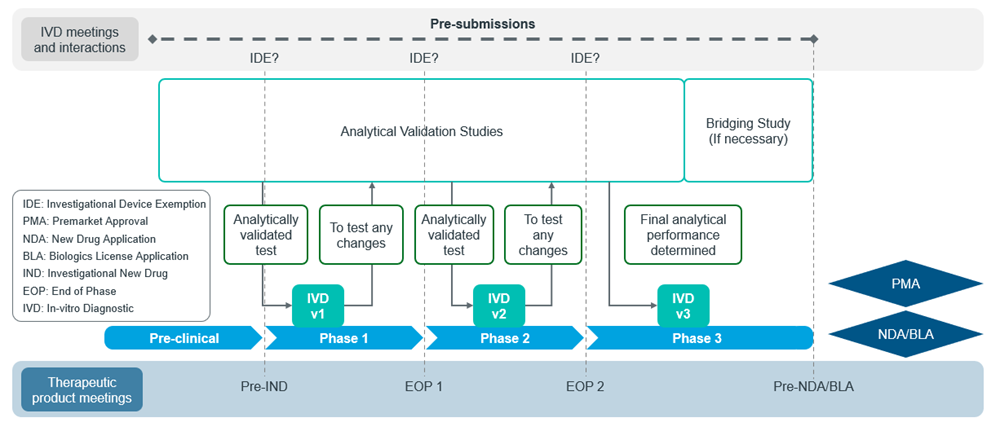
Figure 4: Adapted from US FDA CDx submission process11
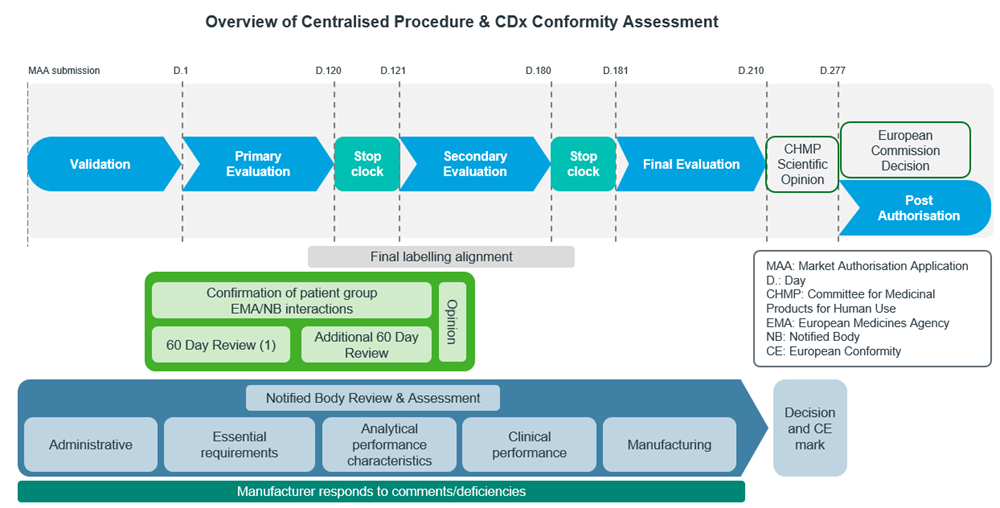
Figure 5: Adapted from EU EMA CDx submission process12
CDx landscape in APAC today
While the discussion so far has been about the progress of CDx regulations in the US and EU, Asia Pacific (APAC) is a market that has been expanding alongside these bigger markets. In fact, the CDx market in APAC is projected to have a CAGR of 13.55% during 2022-2030, with the region having the highest growth rate in the world13, although North America still dominates in terms of market share14.
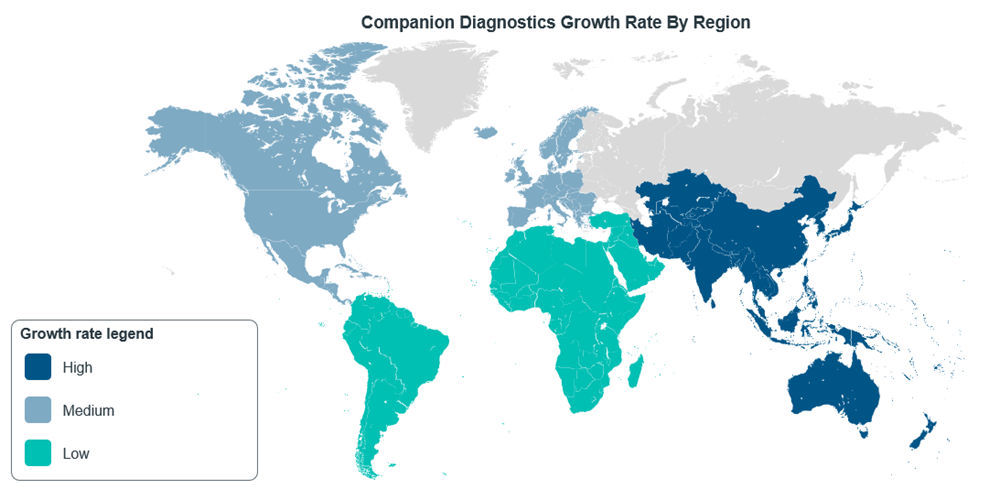
Figure 6: CDxs market growth rate around the world, adapted from Mordor Intelligence14
Despite the general market trend in APAC, countries in the region differ in the extent of regulations in place for CDx, with only South Korea15, Japan16, Australia17 and Taiwan18 publishing established guidelines for CDxs so far.
Although the definition of CDx align with reference agencies in most countries, there are notable differences in availability of CDx-specific guidelines and requirement of concurrent submission of drug and CDx. For a quick cross-country overview, see the table below.
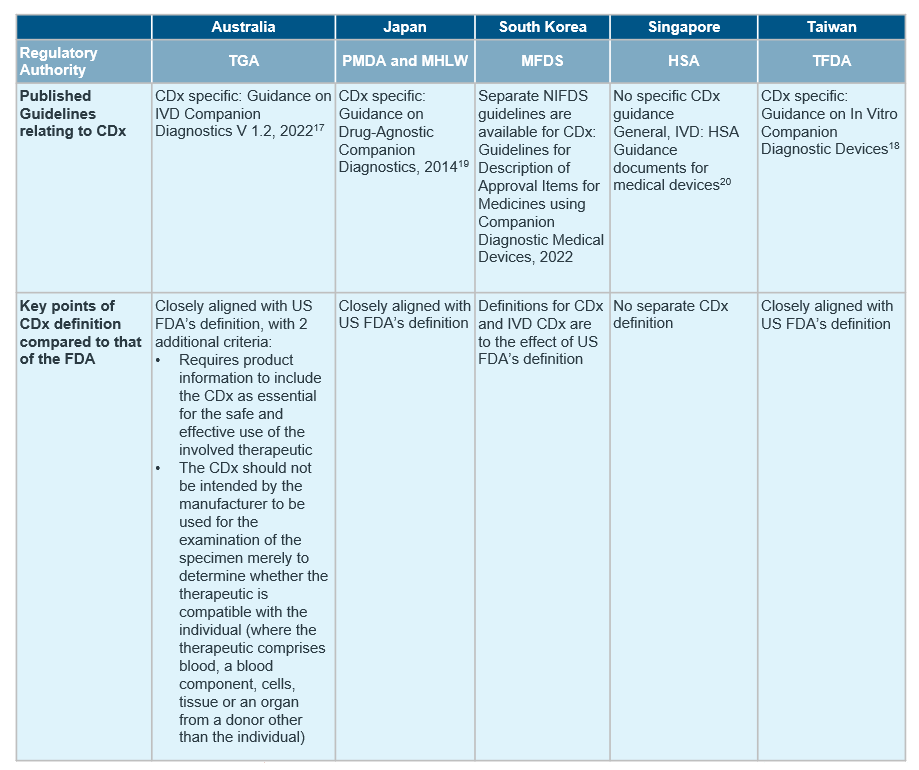
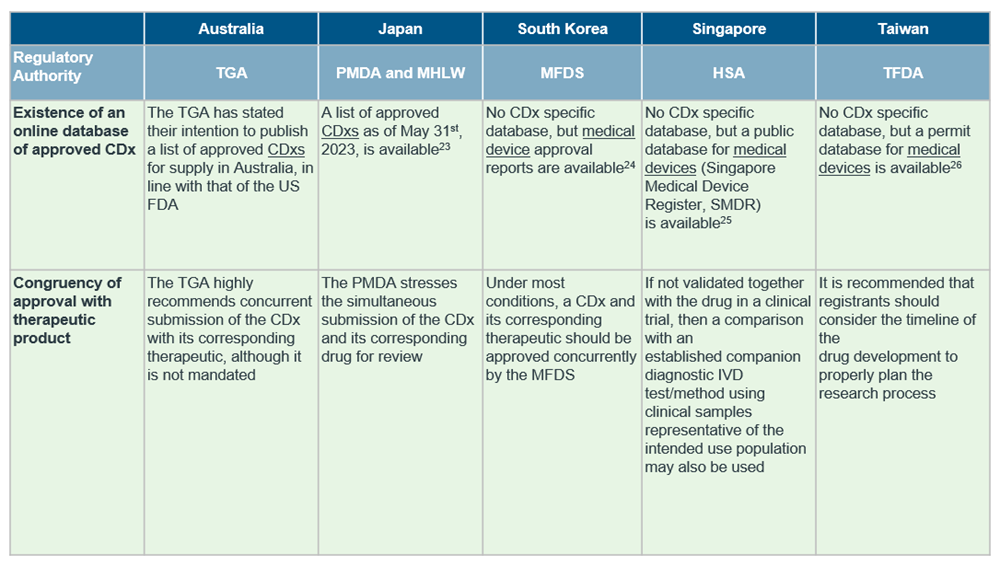
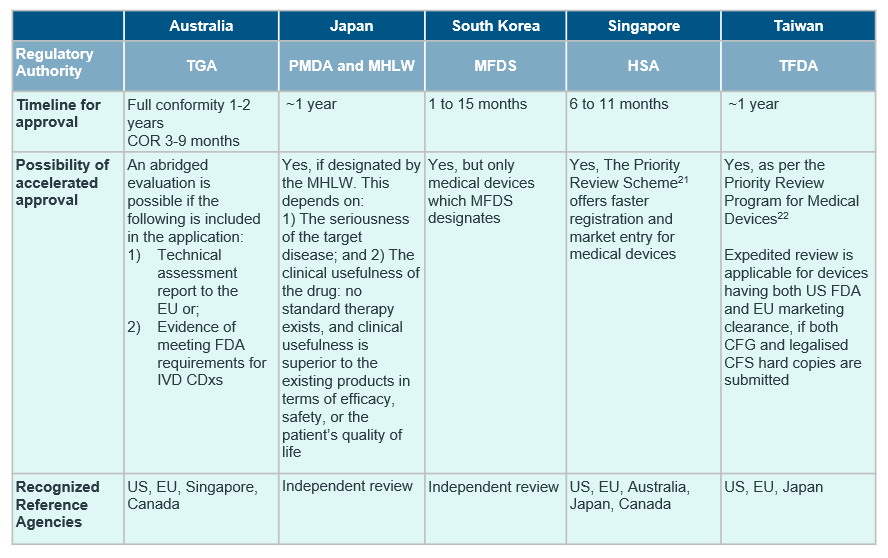
Our recommendations
IQVIA’s experience in CDxs ranges from the design and execution of performance evaluation studies to understanding different regulatory landscapes of countries within the region, as seen in the above table.
From the real-world cases we have worked with, we have observed that companies may try to match the need-for-speed in implementing CDx locally via the lab-developed test (LDT) route, which is usually less tightly regulated and requires less formal registration compared to CDxs -- where there is increased regulatory oversight for technology such as next-generation sequencing.
As such, IQVIA recommends that companies seek to understand country-specific laws and regulations surrounding CDx, such as CDx-specific regulations on technology or involvement of genetic resources. Awareness of the country-specific conditions (e.g., labelling alignment with therapeutic drug) would also help to position the CDx more favourably for reimbursement.
For more information on how CDx is transforming the treatment landscape and improving healthcare outcomes, contact us.
Additional resources
Orellana García, L. P., Ehmann, F., Hines, P. A., Ritzhaupt, A., & Brand, A. (2021). Biomarker and companion diagnostics—a review of medicinal products approved by the European Medicines Agency. Frontiers in Medicine, 8. https://doi.org/10.3389/fmed.2021.753187
U.S. Food and Drug Administration . (n.d.). Laboratory Developed Tests. https://www.fda.gov/medical-devices/in-vitro-diagnostics/laboratory-developed-tests
U.S. Food and Drug Administration . (n.d.). Overview of IVD Regulation. https://www.fda.gov/medical-devices/ivd-regulatory-assistance/overview-ivd-regulation#1
In Vitro Companion Diagnostic Devices . Guidance for Industry and Food and Drug Administration Staff. (2014). https://www.fda.gov/media/81309/download
References
[1] Jain, N. (2019, January 1). Why precision medicine is the future of Healthcare. Technological Transformation. https://www.weforum.org/agenda/2019/01/why-precision-medicine-is-the-future-of-healthcare
[2] Craig, A. (2017). Personalised medicine with companion diagnostics: The intercept of medicines and medical devices in the regulatory landscape. EMJ Innovations, 47–53. https://doi.org/10.33590/emjinnov/10310685
[3] FDA. (n.d.). Companion diagnostics. U.S. Food and Drug Administration. https://www.fda.gov/medical-devices/in-vitro-diagnostics/companion-diagnostics
[4] Jørgensen, J. T. (2021). The current landscape of the FDA approved Companion Diagnostics. Translational Oncology, 14(6). https://doi.org/10.1016/j.tranon.2021.101063
[5] Jørgensen, J. T., Winther, H., Askaa, J., Andresen, L., Olsen, D., & Mollerup, J. (2021). A Companion Diagnostic With Significant Clinical Impact in Treatment of Breast and Gastric Cancer. Frontiers in oncology, 11, 676939. https://doi.org/10.3389/fonc.2021.676939
[6] Martinez, L., Ward, K., Honeywell, M., Thomas, T. A., & Welch , T. (2011, September 20). Herceptest for the detection of HER2 protein overexpression in breast and gastric cancers. Oncology . https://www.uspharmacist.com/article/herceptest-for-the-detection-of-her2-protein-overexpression-in-breast-and-gastric-cancers
[7] American Association for Cancer Research (AACR). (2019, December 27). What is Precision Cancer Medicine? https://www.aacr.org/patients-caregivers/progress-against-cancer/what-is-precision-cancer-medicine/
[8] Mateo, J., Steuten, L., Aftimos, P., André, F., Davies, M., Garralda, E., Geissler, J., Husereau, D., Martinez-Lopez, I., Normanno, N., Reis-Filho, J. S., Stefani, S., Thomas, D. M., Westphalen, C. B., & Voest, E. (2022). Delivering precision oncology to patients with cancer. Nature Medicine, 28(4), 658–665. https://doi.org/10.1038/s41591-022-01717-2
[9] Cheng, Y., Zhang, T., & Xu, Q. (2021). Therapeutic advances in non-small cell lung cancer: Focus on clinical development of targeted therapy and immunotherapy. MedComm, 2(4), 692–729. https://doi.org/10.1002/mco2.105
[10] U.S. Food and Drug Administration. (2020, April). Developing and Labeling In Vitro Companion Diagnostic Devices for a Specific Group of Oncology Therapeutic Products. https://www.fda.gov/media/120340/download
[11] U.S. Food and Drug Administration. (2016, July 15). Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product. https://www.fda.gov/media/99030/download
[12] EFPIA & MedTech Europe. (2020). Determining the path for assessment of a Companion Diagnostic (CDx) under the In Vitro Diagnostic Medical Devices Regulation. https://efpia.eu/media/554434/2020_05_27_efpia-mte_determining-the-path-for-assessment-of-cdx-under-ivdr_final.pdf
[13] Inkwood Research . (2022, August 8). Asia-Pacific Companion Diagnostics Market Forecast 2022-2030. https://inkwoodresearch.com/reports/asia-pacific-companion-diagnostics-market/
[14] Mordor Intelligence. (n.d.). Companion Diagnostics Market Size & Share Analysis - Growth Trends and Forecasts (2023- 2028). https://www.mordorintelligence.com/industry-reports/companion-diagnostics-market
[15] Ministry of Food and Drug Safety . (n.d.). Regulations. Medical Devices. https://www.mfds.go.kr/eng/brd/m_40/list.do
[16] Pharmaceuticals and Medical Devices Agency. (n.d.). Companion diagnostics WG. Companion Diagnostics WG | Pharmaceuticals and Medical Devices Agency. https://www.pmda.go.jp/english/rs-sb-std/rs/0006.html
[17] Therapeutic Goods Administration. (2022, October). IVD Companion Diagnostics Guidance on Regulatory Requirements. https://www.tga.gov.au/sites/default/files/ivd-companion-diagnostics.pdf
[18] Taiwan Food and Drug Administration (2020, July 16). Guidance on In Vitro Companion Diagnostic Devices 伴隨式體外診斷醫療器材技術基準.pdf
[19] Pharmaceuticals and Medical Devices Agency (2022, July 4). Guidance on Drug-Agnostic Companion Diagnostics 000248182.pdf (pmda.go.jp)
[20] Health Sciences Authority . (n.d.). Guidance documents for medical devices. HSA. https://www.hsa.gov.sg/medical-devices/guidance-documents
[21] Health Sciences Authority. (n.d.). Priority Review Scheme. HSA. https://www.hsa.gov.sg/medical-devices/registration/priority-review-scheme
[22] Taiwan Food and Drug Administration. (2017, January 9) Priority Review Program for Medical Devices. https://www.fda.gov.tw/eng/siteList.aspx?sid=10335
[23] Pharmaceuticals and Medical Devices Agency (2023, May 31). List of in vitro Companion Diagnostics or Medical Devices (CDx Products) Approved in Japan. 000246969.pdf (pmda.go.jp)
[24] Ministry of Food and Drug Safety (n.d.) Products. https://www.mfds.go.kr/eng/brd/m_41/list.do
[25] Health Sciences Authority. (n.d.) Public Enquiry – Singapore Medical Devices Register (SMDR). https://eservice.hsa.gov.sg/medics/md/mdEnquiry.do?action=getAllDevices&_ga=2.183810082.563179921.1554083187-551332391.1551944793
[26] Taiwan Food and Drug Administration. (n.d.) https://info.fda.gov.tw/MLMS/H0001.aspx